(Acest articol a fost publicat pentru prima dată pe Rstats – cuantificatși cu amabilitate a contribuit la R-bloggeri). (Puteți raporta problema legată de conținutul acestei pagini aici)
Doriți să vă distribuiți conținutul pe R-bloggeri? dați clic aici dacă aveți un blog, sau aici dacă nu aveți.
am vrut sa stiu:
- câte proteine din proteomul uman au domenii transmembranare?
- dintre cei care au, câți au 1 sau 2 sau n domenii transmembranare?
După puțină căutare, nu am găsit niciun răspuns. Așa că am decis să folosesc R pentru a prelua informațiile necesare de la Uniprot și pentru a le calcula eu. M-am gandit sa il postez aici in caz ca va fi de folos altora.
Uman
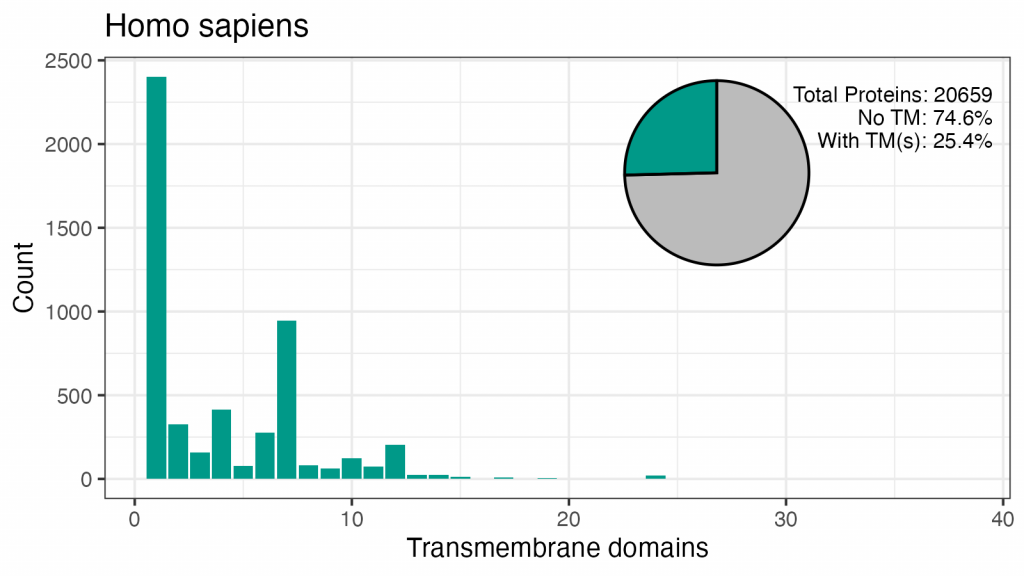
Vom începe cu informațiile pe care le doream. Conform Uniprot, în proteomul uman există 20.659 de proteine. Un sfert dintre aceștia au unul sau mai multe TMD. Majoritatea au un TMD și există aproape 1.000 de proteine 7TM (toate acele GPCR-uri, cred). Există 413 proteine 4TM și 327 2TM. Putem găsi exemple de la 1 la 17 TMD, nu există proteine cu 18, 4 proteine cu 19TM, 21 cu 24TM și 2 cu 38TM.
Analiza se face pur și simplu uitându-se la câte TRANSMEM Uniprot are pentru fiecare ID-uri în proteomul de referință. Nu am făcut distincția între intrările elicoidale și parțiale și, desigur, este posibil ca adnotările să nu fie chiar corecte.
| TMD-uri | Conta | Frecvență (proteomul ca %) | Frecvență (TMDP-uri ca %) |
| 1 | 2402 | 11.6 | 45,8 |
| 2 | 327 | 1.6 | 6.2 |
| 3 | 159 | 0,8 | 3.0 |
| 4 | 413 | 2.0 | 7.9 |
| 5 | 77 | 0,4 | 1.5 |
| 6 | 276 | 1.3 | 5.3 |
| 7 | 947 | 4.6 | 18.0 |
| 8 | 83 | 0,4 | 1.6 |
| 9 | 63 | 0,3 | 1.2 |
| 10 | 123 | 0,6 | 2.3 |
| 11 | 75 | 0,4 | 1.4 |
| 12 | 202 | 1.0 | 3.8 |
| 13 | 24 | 0,1 | 0,5 |
| 14 | 26 | 0,1 | 0,5 |
| 15 | 13 | 0,1 | 0,2 |
| 16 | 1 | 0,0 | 0,0 |
| 17 | 9 | 0,0 | 0,2 |
| 19 | 4 | 0,0 | 0,1 |
| 24 | 21 | 0,1 | 0,4 |
| 38 | 2 | 0,0 | 0,0 |
După ce am scris acest cod, am decis să rulez alți proteomi pentru a vedea cum se compară.
Organisme model
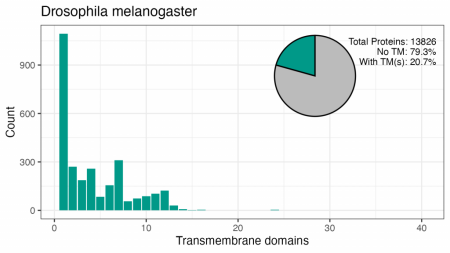
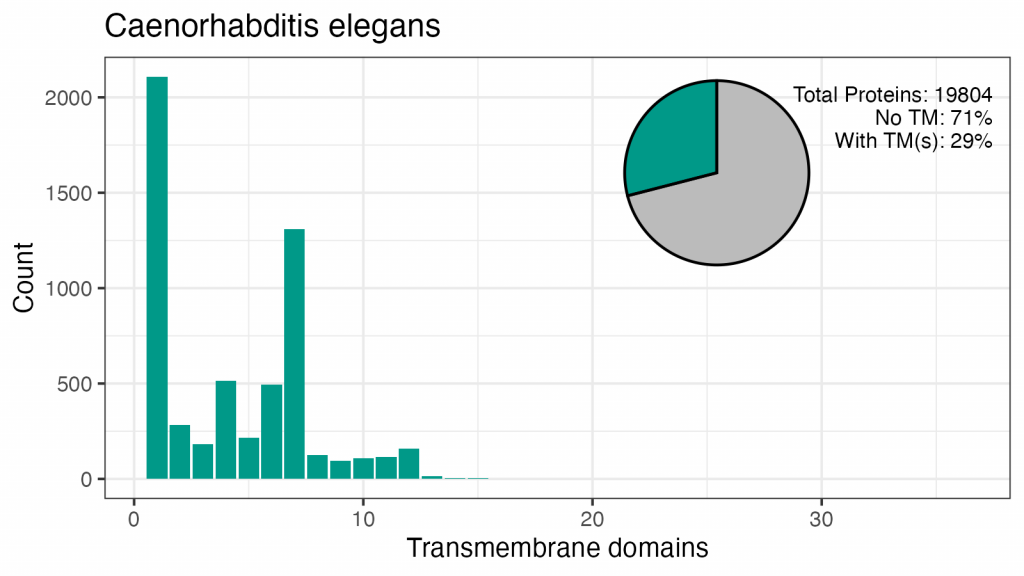
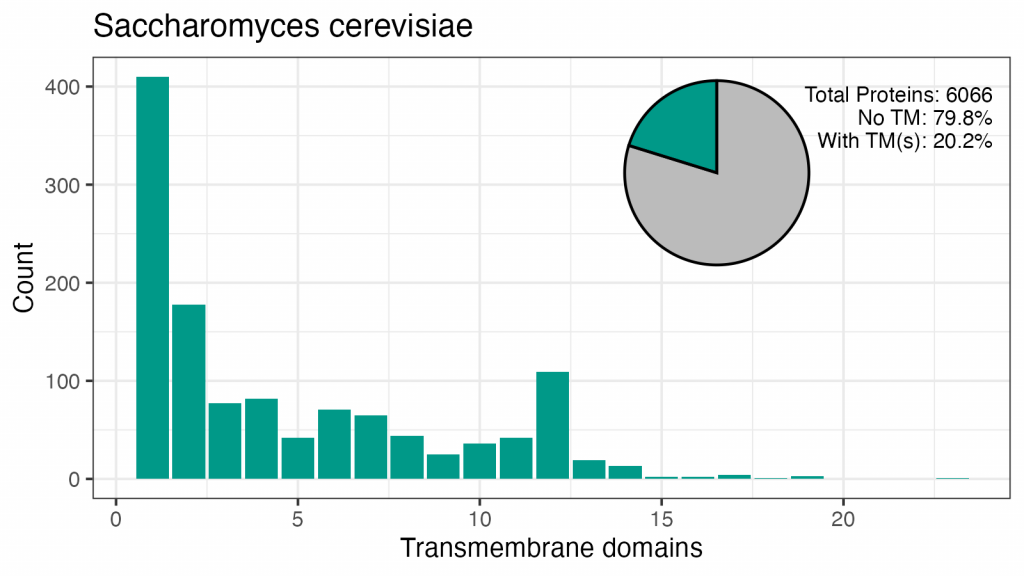
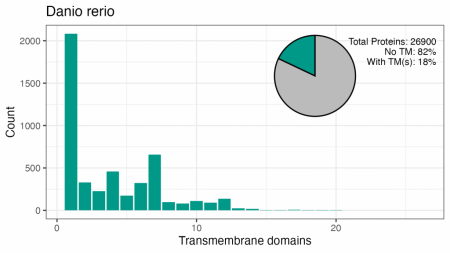
Aceste patru organisme au între 18% și 29% din proteomul format din proteine cu TMD. Modelul numerelor de TMD sunt similare, deși nu există un vârf în drojdie pentru 7TM, iar vârfurile pentru 2, 4, 6 sau 12 TM-uri diferă de cele umane.
Poate că aceste informații sunt acolo într-o bază de date sau alta. După cum am spus, nu am putut găsi ceva ușor. Chiar dacă există modalități mai precise de a determina TMD-urile, cred că aceste date sunt suficient de bune pentru a ști aproximativ care sunt proporțiile.
Codul
Am descărcat manual fișierele fasta.gz pentru proteoamele de referință. Momentan sunt legate aici.
Pentru a extrage toate ID-urile Uniprot, am folosit un shell one-liner:
awk '/^>sp|.*|/{gsub(/^>sp|/,""); gsub(/|.*/,""); print ">" $0; next} {print}' file.fasta | grep "^>" | sed 's/>//g' > species_uniprot.txt
Apoi am folosit acest script R. Funcția principală poate fi probabil simplificată. A trebuit să adaug mai multe verificări pentru a mă asigura că am primit toate datele înapoi de la API. Inainte de a posta, am incercat sa o taiez pentru a fi mai usor de citit, dar I-am reusit doar sa sparg scenariul! Aceasta este versiunea de lucru.
# if (!require("BiocManager", quietly = TRUE)) {
# install.packages("BiocManager")
# }
# BiocManager::install("biomaRt")
library(httr)
library(stringr)
library(ggplot2)
library(biomaRt)
library(dplyr)
library(tidyr)
library(cowplot)
## FUNCTIONS ----
isJobReady <- function(jobId, pollingInterval = 5, maxWaitSeconds = 3600) {
if (is.null(jobId) || length(jobId) == 0 || is.na(jobId) || !nzchar(jobId)) {
return(FALSE)
}
nTries <- ceiling(maxWaitSeconds / pollingInterval)
for (i in 1:nTries) {
url <- paste("https://rest.uniprot.org/idmapping/status/", jobId, sep = "")
r <- GET(url = url, accept_json())
status <- content(r, as = "parsed")
if (!is.null(status(("results"))) || !is.null(status(("failedIds")))) {
return(TRUE)
}
if (!is.null(status(("messages")))) {
print(status(("messages")))
return(FALSE)
}
Sys.sleep(pollingInterval)
}
return(FALSE)
}
retrieveUniprotInfo <- function(x,
chunk_size = 5000,
maxWaitSeconds = 3600,
taxId = "9606",
progress = TRUE) {
normalize_uniprot_ids <- function(values) {
values <- trimws(values)
# Accept FASTA-style headers like: sp|P12345|... or tr|A0A...|...
m <- str_match(values, regex("^>?\s*(?:sp|tr)\|((^|)+)\|", ignore_case = TRUE))
values <- ifelse(!is.na(m(, 2)), m(, 2), values)
values
}
ids <- unique(normalize_uniprot_ids(x))
ids <- ids(!is.na(ids) & nzchar(ids))
if (length(ids) == 0) {
stop("No valid identifiers were provided to retrieveUniprotInfo().")
}
fields <- "accession,id,protein_name,gene_names,ft_transmem,length,cc_function,cc_subcellular_location,go_p,go_c"
acc_pattern <- "^(OPQ)(0-9)(A-Z0-9){3}(0-9)(-(0-9)+)?$|^(A-NR-Z)(0-9)(?:(A-Z)(A-Z0-9){2}(0-9)){1,2}(-(0-9)+)?$"
is_accession <- str_detect(ids, acc_pattern)
split_into_chunks <- function(values, chunk_size = chunk_size) {
split(values, ceiling(seq_along(values) / chunk_size))
}
get_next_link <- function(link_header) {
if (is.null(link_header)) {
return(NULL)
}
links <- unlist(strsplit(link_header, ",\s*"))
next_link <- links(str_detect(links, "rel=\"next\""))
if (length(next_link) == 0) {
return(NULL)
}
next_url <- str_extract(next_link(1), "(?<=<).+?(?=>)")
if (is.na(next_url) || !nzchar(next_url)) {
return(NULL)
}
next_url
}
read_tsv_response <- function(resp) {
read.table(
text = content(resp, as = "text", encoding = "UTF-8"),
sep = "t",
header = TRUE,
fill = TRUE,
quote = "",
comment.char = "",
check.names = FALSE
)
}
fetch_from_redirect <- function(redirect_url) {
if (is.null(redirect_url) || length(redirect_url) == 0 ||
is.na(redirect_url) || !nzchar(redirect_url)) {
return(NULL)
}
# The paged idmapping results endpoint is capped at size <= 500.
# Use stream endpoint to retrieve the full chunk in one response.
stream_url <- gsub("/results/", "/results/stream/", redirect_url)
sep <- ifelse(str_detect(stream_url, "\?"), "&", "?")
stream_url <- paste0(
stream_url,
sep,
"fields=", URLencode(fields, reserved = TRUE),
"&format=tsv"
)
r <- GET(url = stream_url)
if (status_code(r) < 400) {
return(read_tsv_response(r))
}
# Fallback to paged endpoint if stream is unavailable.
sep <- ifelse(str_detect(redirect_url, "\?"), "&", "?")
url <- paste0(
redirect_url,
sep,
"fields=", URLencode(fields, reserved = TRUE),
"&format=tsv&size=500"
)
r <- GET(url = url)
stop_for_status(r)
resultsTable <- read_tsv_response(r)
next_url <- get_next_link(headers(r)(("link")))
while (!is.null(next_url) && !is.na(next_url) && nzchar(next_url)) {
r <- GET(url = next_url)
stop_for_status(r)
resultsTable <- rbind(resultsTable, read_tsv_response(r))
next_url <- get_next_link(headers(r)(("link")))
}
resultsTable
}
map_ids <- function(values, from_db, to_db, chunk_size, taxId = NULL,
label = "ids") {
if (length(values) == 0) {
return(NULL)
}
results_list <- list()
chunks <- split_into_chunks(values, chunk_size = chunk_size)
n_chunks <- length(chunks)
for (i in seq_along(chunks)) {
chunk <- chunks((i))
if (isTRUE(progress)) {
cat(sprintf("(UniProt) %s chunk %d/%d (%d ids)n",
label, i, n_chunks, length(chunk)))
}
files <- list(
ids = paste0(chunk, collapse = ","),
from = from_db,
to = to_db
)
if (!is.null(taxId)) {
files$taxId <- taxId
}
r <- POST(url = "https://rest.uniprot.org/idmapping/run", body = files,
encode = "multipart", accept_json())
stop_for_status(r)
submission <- content(r, as = "parsed", encoding = "UTF-8")
job_id <- submission(("jobId"))
if (is.null(job_id) || length(job_id) == 0 || is.na(job_id) || !nzchar(job_id)) {
if (isTRUE(progress)) {
cat(sprintf("(UniProt) %s chunk %d/%d: no jobId returnedn",
label, i, n_chunks))
}
next
}
if (!isJobReady(job_id, maxWaitSeconds = maxWaitSeconds)) {
if (isTRUE(progress)) {
cat(sprintf("(UniProt) %s chunk %d/%d: timeout/not readyn",
label, i, n_chunks))
}
next
}
details_url <- paste("https://rest.uniprot.org/idmapping/details/",
job_id, sep = "")
r <- GET(url = details_url, accept_json())
stop_for_status(r)
details <- content(r, as = "parsed", encoding = "UTF-8")
redirect_url <- details(("redirectURL"))
if (is.null(redirect_url) || length(redirect_url) == 0 ||
is.na(redirect_url) || !nzchar(redirect_url)) {
if (isTRUE(progress)) {
cat(sprintf("(UniProt) %s chunk %d/%d: missing redirectURLn",
label, i, n_chunks))
}
next
}
chunk_result <- fetch_from_redirect(redirect_url)
if (is.null(chunk_result)) {
if (isTRUE(progress)) {
cat(sprintf("(UniProt) %s chunk %d/%d: invalid redirectURLn",
label, i, n_chunks))
}
next
}
results_list((length(results_list) + 1)) <- chunk_result
if (isTRUE(progress)) {
cat(sprintf("(UniProt) %s chunk %d/%d: completedn",
label, i, n_chunks))
}
}
if (length(results_list) == 0) {
return(NULL)
}
do.call(rbind, results_list)
}
accession_ids <- ids(is_accession)
gene_like_ids <- ids(!is_accession)
acc_results <- map_ids(
values = accession_ids,
from_db = "UniProtKB_AC-ID",
to_db = "UniProtKB",
chunk_size = chunk_size,
label = "accessions"
)
gene_results <- map_ids(
values = gene_like_ids,
from_db = "Gene_Name",
to_db = "UniProtKB-Swiss-Prot",
chunk_size = chunk_size,
taxId = taxId,
label = "gene_names"
)
results_list <- list()
if (!is.null(acc_results)) {
results_list((length(results_list) + 1)) <- acc_results
}
if (!is.null(gene_results)) {
results_list((length(results_list) + 1)) <- gene_results
}
if (length(results_list) == 0) {
stop("No UniProt results were returned. Check identifiers and taxId.")
}
resultsTable <- do.call(rbind, results_list)
if ("Entry" %in% colnames(resultsTable)) {
resultsTable <- resultsTable(!duplicated(resultsTable$Entry), )
}
return(resultsTable)
}
## SCRIPT ----
species <- c("human", "zebrafish", "drosophila", "worm", "yeast")
sci_names <- c("human" = "Homo sapiens", "zebrafish" = "Danio rerio", "drosophila" = "Drosophila melanogaster",
"worm" = "Caenorhabditis elegans", "yeast" = "Saccharomyces cerevisiae")
for (org in species) {
# look up scientific name of org
sci_name <- sci_names(org)
output_path <- paste0("Output/Data/", org, "_uniprot.csv")
if(file.exists(output_path)) {
message(paste("File", output_path, "already exists. Loading", org))
df <- read.csv(output_path)
} else {
message(paste("Retrieving UniProt info for", org))
species_ids <- read.delim(paste0("Data/", org, "_uniprot.txt"), header = FALSE)
names(species_ids) <- c("uniprot_id")
df <- retrieveUniprotInfo(species_ids$uniprot_id)
# save this result
write.csv(df, output_path, row.names = FALSE)
}
df$tms <- str_count(df$Transmembrane, "TRANSMEM")
tm_counts <- df %>%
group_by(tms) %>%
summarise(count = n()) %>%
filter(tms > 0)
p1 <- ggplot(tm_counts, aes(x = tms, y = count)) +
geom_col(fill = "#009988") +
labs(x = "Transmembrane domains", y = "Count",
title = sci_name) +
lims(x = c(0.5,NA), y = c(0,NA)) +
theme_bw(10)
p2 <- SuperPlotR::pieplot(x1 = c(sum(df$tms == 0), sum(df$tms > 0)),
cols = c("#bbbbbb", "#009988")) +
# blank background and no legend
theme_void() +
theme(legend.position = "none")
# inset p2 in p1 and add information about the percentages
p <- ggdraw() +
draw_plot(p1) +
# top right
draw_plot(p2, x = 0.9, y = 0.9, hjust = 1, vjust = 1, width = 0.4, height = 0.4) +
draw_label(paste0("Total Proteins: ", nrow(df),
"nNo TM: ", round(sum(df$tms == 0) / nrow(df) * 100, 1),
"%nWith TM(s): ",
round(sum(df$tms > 0) / nrow(df) * 100, 1), "%"),
x = 0.97, y = 0.85, hjust = 1, vjust = 1, size = 8)
plot_path <- paste0("Output/Plots/", org, "_uniprot_tm_counts.png")
ggsave(plot_path, p, width = 1600, height = 900, units = "px", dpi = 300)
}
Rețineți că folosesc {cowplot} la sfârșit pentru a introduce diagrama circulară și pentru a adăuga textul pe diagrama principală.
—
Titlul postării provine din „Domeniul meu” de Bernard Butler de pe albumul său People Move On.